
Pharmacokinetic models describe the rate of absorption, distribution, metabolism, and elimination of a particular drug. This can vary significantly based on properties of the drug itself, varying levels of metabolism in the context of organ impairment or enzyme polymorphisms, plasma protein binding, and other patient factors (ie, obesity).
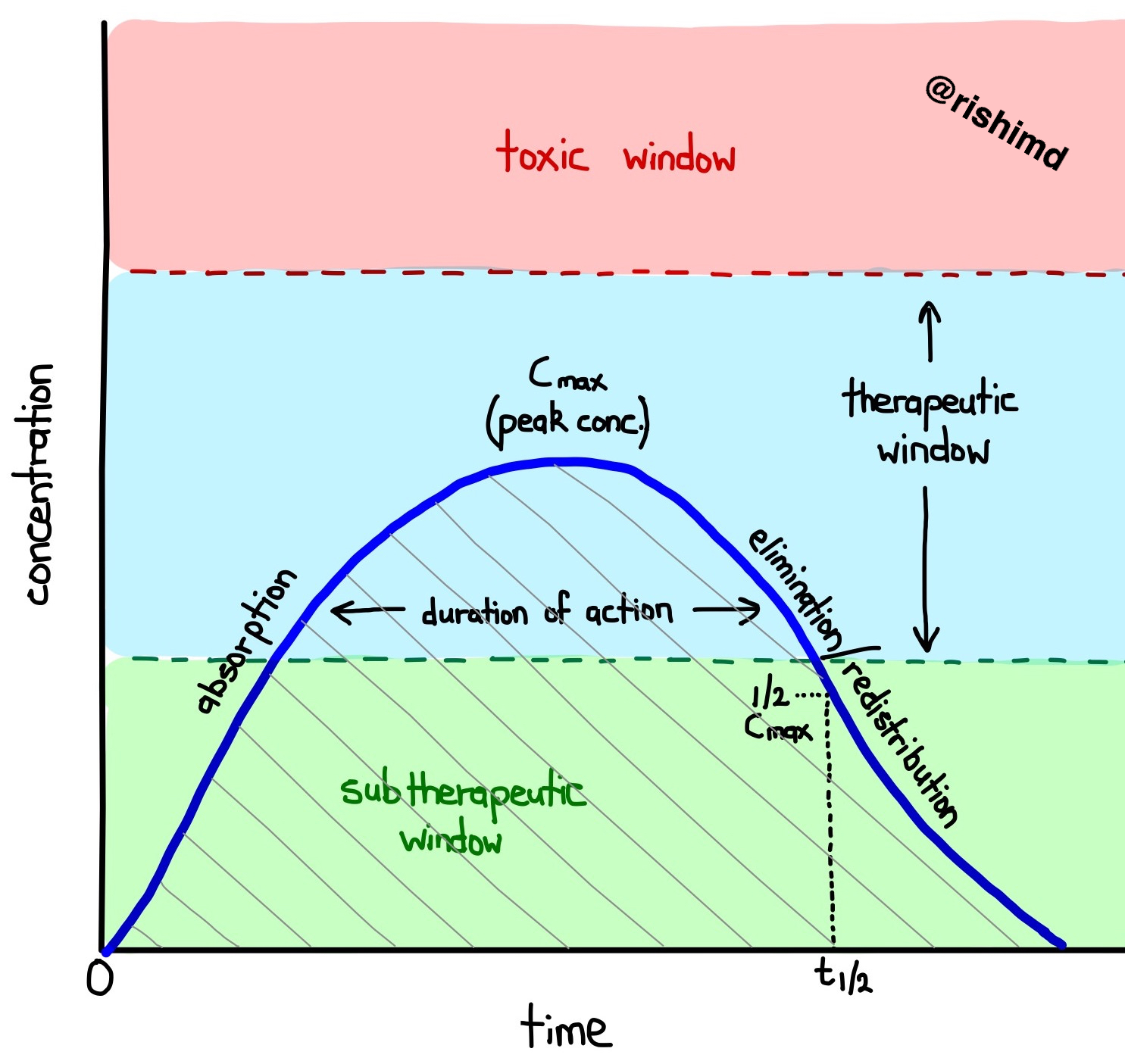
When a drug is injected intravenously at time 0, it is 100% bioavailable and undergoes absorption into various tissues and fluid compartments. Its concentration at the site of action rises to a therapeutic window allowing the drug to exert its clinical effect. Although some drugs may be considered “short acting”, they might not be entirely eliminated from the body (ie, the concentration might just be in the subtherapeutic window).
Based on the initial dose, a “maximum plasma concentration” (Cmax) is reached with the goal of avoiding the drug’s toxic window. The time at which half this concentration is achieved is the drug’s effective half life (t1/2) although most drugs have several elimination half lives (a more complex topic). The duration of action describes how long the drug’s concentration at its effector site is in the therapeutic window.
Understanding these models explains why certain medications have specific loading doses, dosing intervals, etc. I’ve admittedly kept this explanation overly simplified, but the point is to illustrate some of the parameters at play.
Drop me a comment with your thoughts and questions!


