
There are four classic disturbances in traditional acid-base equilibria: respiratory acidosis, respiratory alkalosis, metabolic acidosis, and metabolic alkalosis. The body tries to compensate for a primary disturbance(s) (i.e., metabolic alkalosis) through one or more mechanisms (i.e., respiratory acidosis). For simplicity, respiratory events deal with the lungs and carbon dioxide (pCO2), whereas metabolic events involve the kidneys and bicarbonate (HCO3–). Although the normal values for pCO2 and HCO3– are roughly 40 mmHg and 24 mEq/L, respectively, remember that not all patients live at these values (COPD, chronic diuretic use, etc.). As a side note, remember the [HCO3–] value obtained from an arterial blood gas (ABG) is calculated. When performing detailed acid-base assessments, use a metabolic profile (BMP, CMP) to determine the measured [HCO3-]. Finally, acid-base disturbances must be considered relative to the patient’s baseline! I’ll discuss specific numbers when I talk about “Interpreting A Blood Gas” later in this post.
ANION GAP (AG) METABOLIC ACIDOSIS
So first, let’s talk about metabolic acidosis, which is broken down into anion-gap (AG) metabolic acidosis and non-anion-gap metabolic acidosis (NAGMA)
AG = [Na+] – ([HCO3–] + [Cl–])
Of note, [K+] is omitted due to low plasma concentrations (some of my colleagues overseas include potassium in the calculation). AG metabolic acidosis has a myriad of causes I remember with the mnemonic GOLDMARK:
- G – glycols (methylene and propylene)
- O – oxoproline (metabolite of Tylenol)
- L – L-lactate (isoniazid can increase lactate)
- D – D-lactate
- M – methanol
- A – aspirin/salicylates
- R – renal failure (uremia)
- K – ketoacidosis
The AG expresses the difference between unmeasured cations (Ca2+, Mg2+, etc.) and unmeasured anions (albumin, PO4-3, SO4-2, etc.). A normal AG is 10-14 mEq/L, but keep in mind that the AG decreases by 2.5 mEq/L for every 1 g/dL decrease in albumin (the most abundant, unmeasured anion) from normal (~ 4 g/dL).
NON ANION GAP METABOLIC ACIDOSIS (NAGMA)
Based on the anion gap equation above, a non-anion gap metabolic acidosis (NAGMA) can be thought of as a decrease in bicarbonate being matched by an increase in chloride to maintain the gap. When do we see this situation clinically?
- Saline administration
- Bicarbonate loss through the GI tract (i.e., diarrhea, ileal diversions, pancreatic fistula)
- Renal tubular acidosis (RTA)
- Medications like carbonic anhydrase inhibitors (acetazolamide), Amphotericin B, cyclosporine, etc
METABOLIC ALKALOSIS
Whenever I think about metabolic alkalosis, I look for pathology that causes a loss/shift of H+ (acid) and/or retention of HCO3– (base). I then look at the urine chloride concentration to help determine the etiology.
For example, vomiting leads to a significant loss of H+, K+, and Cl- (or excessive oro/nasogastric tube decompression). This volume loss causes the distal nephron to retain sodium by wasting H+ ions. Let’s apply volume loss to another classic example – “contraction alkalosis.” Decreased extracellular volume (i.e., dehydration) ramps up the renin-angiotensin-aldosterone (RAS) pathway with the net effect of sodium and free water retention with potassium excretion. This increase in tubular potassium is exchanged distally in the kidney (intercalated cells) for H+, thereby acidifying the urine. There we go – another example of H+ loss. In both of the examples above, urine chloride levels should be low (< 25 mEq/L), termed “chloride-responsive” metabolic alkalosis.
When urine chloride is high (> 40 mEq/L), termed “chloride resistant” metabolic alkalosis, I think of conditions where bicarbonate is retained – Bartter syndrome (a defect that acts like a loop diuretic), Gitelman syndrome (a defect that acts like a thiazide diuretic), congenital adrenal hyperplasia from 11β-hydroxylase deficiency, etc.
RESPIRATORY ACIDOSIS
Keep things simple! When CO2 production exceeds CO2 elimination via the lungs in the context of acidosis, you should be worried about respiratory acidosis. I think about anything that can affect the lungs’ ability to ventilate adequately:
- Excessive sedation
- Neuromuscular weakness (myasthenia, Guillain-Barre), deconditioning, electrolyte abnormalities (i.e., hypophosphatemia), malnutrition, splinting
- Intrinsic lung diseases, including ARDS, pneumonia, COPD, pulmonary edema, etc.
RESPIRATORY ALKALOSIS
To a large degree, this can be thought of as the opposite of respiratory acidosis. That is, a condition where CO2 elimination exceeds CO2 production. Think of situations where the minute ventilation (respiratory rate x tidal volume) is increased.
- Anxiety, rapid, shallow breathing
- Salicylate toxicity (stimulates the respiratory center)
- Pregnancy (progesterone stimulates the respiratory center)

INTERPRETING THE ABG
Use the table below to help you answer these questions!
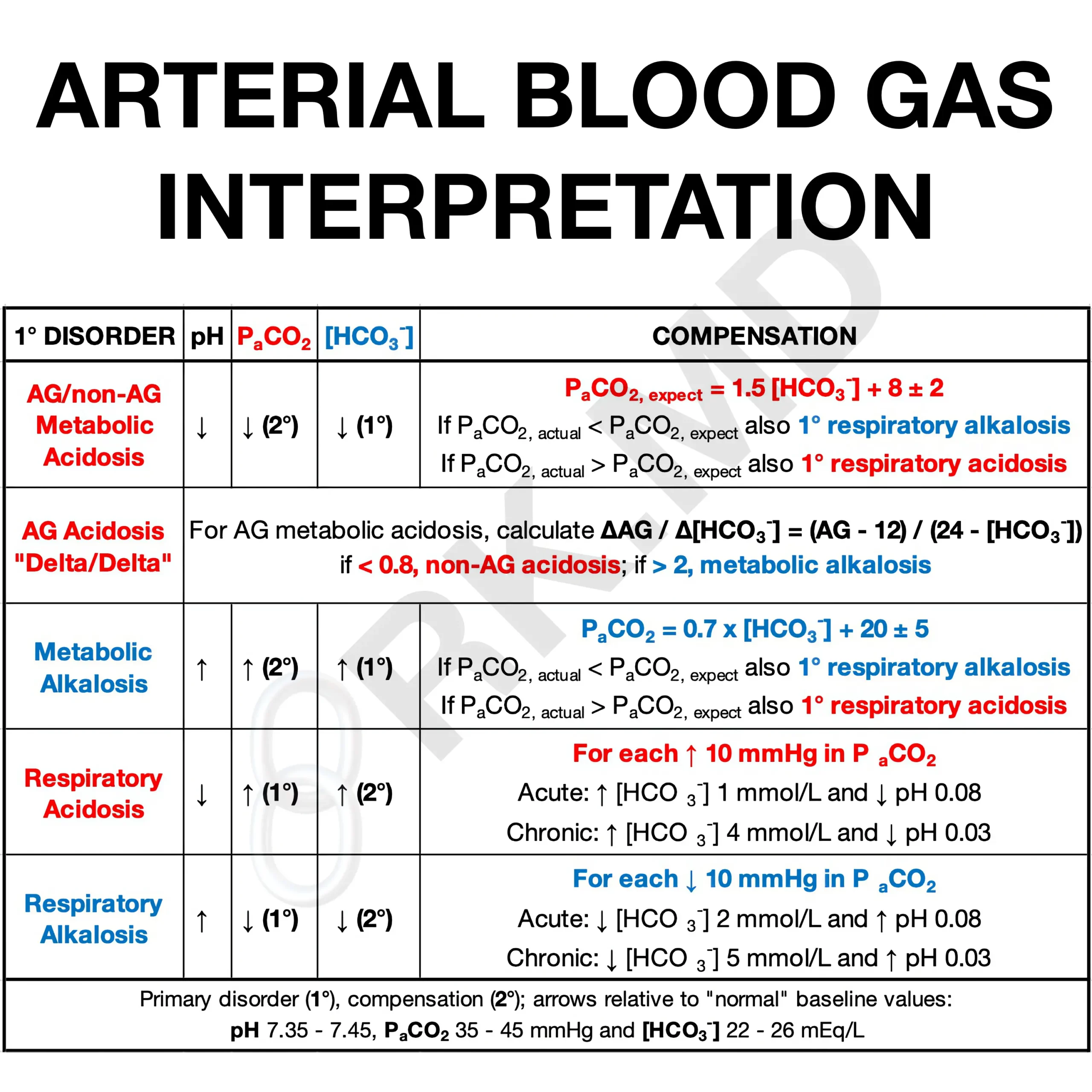
- Is the ABG pH acidotic (< 7.35) or alkalotic (> 7.45)?
- Is the primary disturbance respiratory or metabolic based on PaCO2 and [HCO3–], respectively.
- For metabolic acidosis, calculate the corrected anion gap. For respiratory disturbances, determine if they are acute or chronic based on the pH and PaCO2.
- For metabolic disturbances, is there an appropriate respiratory compensation at play based on Winter’s formula (PaCO2,expect = 1.5 x [HCO3–] + 8 ± 2). Perhaps there is an additional respiratory disturbance superimposed on the compensation?
- If there’s an AG metabolic acidosis, calculate the “delta/delta” (ΔAG / Δ[HCO3–]). This is based on the idea that an increase in AG should be met by a proportionate decrease in [HCO3–], so the ratio will be 1.0. The delta/delta is usually between 1.0 and 2.0 for a pure AG metabolic acidosis. This range is due to the volume of distribution varying among anions and ongoing physiologic buffering. If the delta/delta ratio is ~ 0.8 or less, this suggests another process consuming [HCO3–] like a superimposed NAGMA. If the ratio is > 2, this suggests [HCO3–] is higher than explained by just an AG metabolic acidosis… like a superimposed metabolic alkalosis.
This is a complex yet incredibly important topic in medicine, so please leave me comments with questions below! 🙂



Thanks. Mastering ABGs require frequent practice, I believe. How do you calculate if there is a metabolic derangement e.g. metabolic acidosis if the primary disturbance is respiratory acidosis? Saw an ABG with pH 7.025, pCO2 65mmHg and HCO3 24, anion gap was wide
I do not unterstand the image of the normal abg. Why is it unacceptable? Why are lactate and base excess circled?
Please read the caption below the image – I was just messing with my resident. It’s a normal ABG.
ah ok, sorry XD thank you for these articles. very helpful.
No worries!
When the RAS system kicks in during dehydration, sodium is retained, potassium is secreted, and then potassium is then pushed back into the blood in exchange for hydrogen to pushed into the urine?
Yes, aldosterone promotes sodium absorption and kaluresis (potassium excretion) in the principal cells of the collecting duct epithelium. Intercalated cells (also in the collecting duct) have an H+/K+ ATP-ase that actively absorbs potassium and promotes the excretion of acid (H+).
If Po2 and pc2 both high with low Ph in Koch, s Pt. What’s interpretation n management. Pt was admitted in comatose condition, with H/O loose motions followed by sudden LOC. CBC, LFT, KFT, ELECTROLYTES were normal including RBS
I don’t give medical advice on the Internet. I’m sorry.
Magnificent explanation
Appreciate the feedback!
Thanks a lot for the comprehensive explanation, I was actually preparing the same topic for PICU nurse and it really helped me ???
You’re very welcome! 🙂
This is awesome, Rishi, thank you. We were just discussing non-anion gap metabolic acidosis in rounds last week. I’m a firm believer that if you can’t explain a topic simply, then you don’t understand it well enough. Your explanations are always great.
Thanks so much Tyler! Appreciate the comment!
its v helpful ??!plz upload more on such complex topics
Thanks so much, Alvin! Glad you found it helpful!